"Una famiglia che beve molto"
Una paziente di 48 anni giunge alla nostra attenzione in ambulatorio per “poliuria e polidipsia”.
Dall’anamnesi fisiologica e patologica personale emergono: menarca a 14 anni, 2 gravidanze a termine (2 figli di 11 e 8 anni in abs), in entrambi i casi diabete mellito gestazionale, successivamente glicemia sempre nella norma, anche a recente controllo. Cicli mestruali tuttora regolari, non fuma, non beve alcolici. Dieta varia, moderata attività fisica. BMI 22 kg/m2. Non assume o ha assunto in modo continuativo farmaci. Non patologie di rilievo in passato, non interventi chirurgici, no traumi maggiori. Lavora come architetto, accede volentieri al colloquio.
Riguardo il problema specifico riferisce polidipsia dall’età di 8 anni. Ha sempre bevuto dai 6 ai 9 l di liquidi nelle 24 ore con diuresi sostanzialmente in bilancio con le entrate. Frequenti risvegli notturni per nicturia e necessità di bere. Ha fatto presente in più di un’occasione al proprio medico questa situazione, ma riferisce che il medico ha risposto dicendo che “finchè beve il problema non sussiste”.
Solo dopo l’ennesima segnalazione al medico curante, è stata inviata a visita specialistica endocrinologica. Su richiesta dell’endocrinologo, racconta che anche diversi familiari (sorella, madre, nonno materno, zio e cugina sempre dal ramo materno) bevono molto (tutti almeno 5 litri di liquidi al giorno) e che l’insorgenza della polidipsia e poliuria in tutti i casi è avvenuta tra i 5 e gli 8 anni. La paziente riferisce le lamentele vivaci della nonna materna, quando lei i familiari sono a pranzo da lei; “la nonna dice che siamo tutti pazzi a bere così tanto!”. Nessun familiare è affetto da diabete mellito. Non nota familiarità per altre malattie.
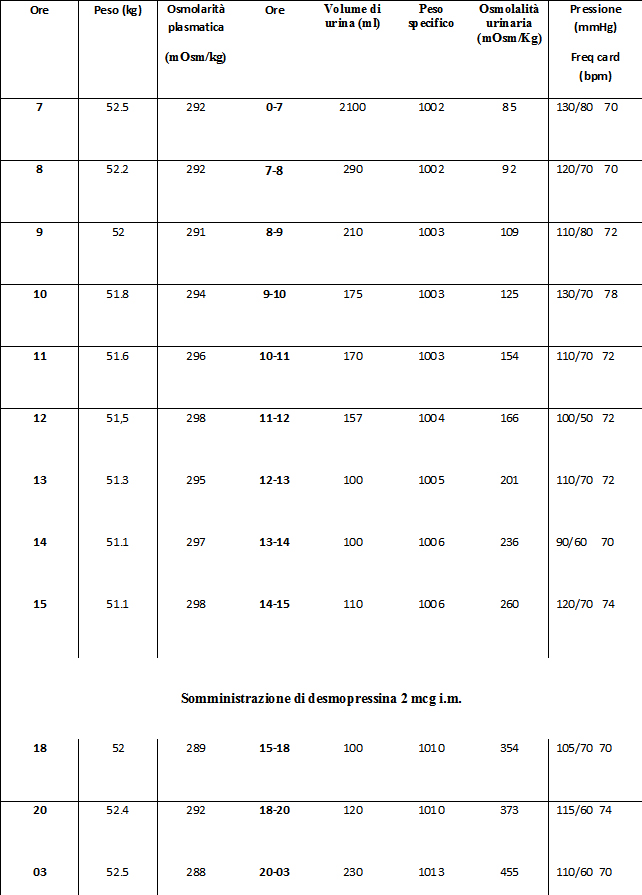
La paziente viene ricoverata per essere sottoposta a test di assetamento + test con desmopressina nel sospetto di diabete insipido (DI).
E’ stata successivamente effettuata anche RMN encefalo, che ha evidenziato ipofisi di dimensioni regolari, peduncolo in asse, non ispessito. Perdita del segnale iperintenso in corrispondenza della neuroipofisi. Non lesioni espansive.
Risposte
1) Sindrome di Wolfram2) DI familiare neurogeno3) DI familiare nefrogeno
4) DI secondario ad ipofisite autoimmune
La risposta corretta è la risposta numero:
2
Risposta Corretta Nr. 2
DI familiare neurogeno
I risultati del test di assetamento + la successiva somministrazione di desmporessina dimostrano inequivocabilmente che la paziente è affetta da
DI neurogeno (parziale concentrazione urinaria durante la fase di assetamento, 260 mOsm/kg, ed aumento dell’osmolarità urinaria, dopo somministrazione di desmopressina, del 75% (455 mOsm/kg) (1).
Considerata la ricorrente presenza di polidipsia/poliuria in famiglia nel ramo materno, il sospetto che si tratti di forma di
DI neurogeno geneticamente determinato è molto elevato (2). Queste forme, associate a mutazioni del gene AVP (20p12), sono molto rare e sono trasmesse nella quasi totalità dei casi con modalità autosomica dominante, che è in accordo con quanto emerge dalla storia della paziente e dei suoi familiari. Di norma la malattia non è presente alla nascita, ma si sviluppa precocemente durante l’infanzia. La maggior parte delle mutazioni identificate è localizzata nella regione del gene che codifica per la proteina di trasporto neurofisina II (NPII). Per effetto della mutazione si ha riarrangiamento della struttura tridimensionale del pro-ormone, che porta ad un suo accumulo intracellulare. Ciò determina citotossicità con danno progressivo, che si manifesta infine con l’insorgenza di DI alcuni anni dopo la nascita (2). Il caso in esame è giunto alla nostra attenzione molto recentemente ed ancora l’analisi genetica non è stata effettuata. Ovviamente, sarà importante valutare, con il loro consenso, anche i familiari “clinicamente” affetti.
La
sindrome di Wolfram è una patologia rara (prevalenza 1:160000-770000) (3). Oltre al DI neurogeno, comuni manifestazioni includono diabete mellito, atrofia del nervo ottico e sordità neurosensoriale. La prima patologia a manifestarsi , attorno ai 6 anni, è di norma il diabete mellito, seguita da atrofia del nervo ottico (perdita della visione dei colori e della visione periferica) attorno agli 11 anni. Più della metà dei pazienti sviluppano ipoacusia che può progressivamente condurre a sordità. Il DI è presente in circa il 70% dei casi. I pazienti possono essere affetti anche da altre alterazioni di tipo neurologico ed urinario. Molti pazienti muoiono prematuramente e l’età media è di circa 30 anni. La conferma della diagnosi clinica è affidata a test genetico per la ricerca di mutazioni del gene WFS1 (4p16.1), che codifica per la Wolframina, proteina localizzata nel reticolo endoplasmico, che svolge un ruolo nell'omeostasi del calcio. La malattia si trasmette nella maggior parte dei casi con modalità recessiva, ma talora anche con modalità dominante. Sono stati descritti anche rari casi di malattia associata a mutazioni di un altro gene, ZCD2 (o WFS2), un gene “zinc finger” altamente conservato (4q22-q24), che codifica per una piccola proteina intermembrana del reticolo endoplasmico. E’ evidente che la storia clinica della paziente e dei suoi familiari non sono indicativi per sindrome di Wolfram, così come non sono suggestivi di un’ipotetica diagnosi di
ipofisite autoimmune. Questa entità nosologica, la cui diagnosi è ancora spesso problematica, è più frequente nel sesso femminile (5:1) e compare più frequentemente nell’ultimo trimestre di gravidanza e nel periodo post-partum. Nella donna l’età media alla diagnosi è di 35 anni circa, 45 anni nell’uomo. Può essere distinta in tre forme, adenoipofisite linfocitaria, infundibulo-neuroipoifisite linfocitaria o panipofisite linfocitaria (4). Clinicamente
si riconosce una fase acuta/subacuta ed una fase cronica. Nella prima fase tipicamente sono presenti sintomi e segni da compressione (es. cefalea, deficit campo visivo), accompagnati da uno o più deficit ipofisari (più freqentemente deficit isolato di ACTH) ed iperprolattinemia. Nella fase cronica la fibrosi ed atrofia ipofisaria risultano in deficit ipofisari. La localizzazione infundibulo-neuroipofisaria può determinare l’insorgenza di DI. Alla RMN l’ipofisi è di norma aumentata di volume, con estensione soprasellare, il peduncolo ipofisario appare frequentemente ispessito (>4 mm), ma non deviato. Se il processo infiammatorio coinvolge la neuroipofisi vi è di solito perdita della tipica iperintensità di segnale.
Bibliografia di riferimento
- Robertson GL 1995 Diabetes insipidus. Endocrinol Metab Clin North Am 24:549-572
- Baglioni S, Corona G, Maggi M, Serio M, Peri A 2004 Identification of a novel mutation in the AVP-NPII gene affecting the sixth intrachain disulfide bond of the neurophysin II moiety. Eur. J. Endocrinol. 151:605-611
- Urano F 2016 Wolfram Syndrome: Diagnosis, Management, and Treatment.Curr Diab Rep. 16:6.
- Bellastella G, Maiorino MI, Bizzarro A, Giugliano D, Esposito K, Bellastella A, De Bellis A 2016 Revisitation of autoimmune hypophysitis: knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary 19:625-642
Siamo spiacenti, non è possibile rispondere al questionario in questo momento.






